| ||||||||||
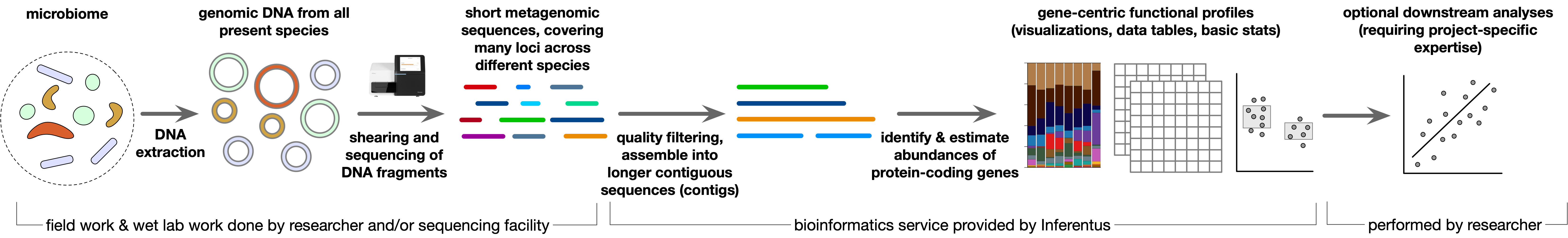
Gene-centric metagenomicsCode: METAGC01 Short-read gene-centric metagenomics, including contig assembly, detection and functional annotation of protein-coding sequences, gene abundance profiles (KEGG), pairwise dissimilarity metrics, MDS and various visualizations. The client provides raw shotgun metagenomic sequences and sample metadata. Log in to see availability and payment modalities. Metagenomic sequencing is a well-established and powerful technique in modern microbiology, in which random DNA fragments obtained from a biological sample are sequenced and analyzed. These DNA fragments generally originate from many different taxa and from all genomic regions, thus offering a deep view of a microbial community. Metagenomics has been used to study microbial communities in virtually all ecosystems, ranging from the deep ocean and subsurface sediments, to soil, bioreactors and the human gut. In gene-centric metagenomics, one specifically focuses on the detection and identification of protein-coding genes, or fragments of such genes, in order to determine the likely biochemical functions or metabolic potential of resident microbes. This information becomes very powerful when combined with experimental treatments, or surveys across space or time. For example, one can use gene-centric metagenomics to determine the proportions of microbes capable of photosynthesis, iron respiration or denitrification, or the prevalence of antibiotic resistance genes, separately in each sample. One could then examine whether these variables change across an environmental gradient, or between patient treatment groups. It is important to note that these "functional profiles" summarize the overall microbial community in each sample, and do not resolve which taxon happens to encode which biochemical function. For many studies this is plenty and sufficient information. If an identification of individual taxa involved with specific biochemical functions is needed, one can instead use genome-resolved metagenomics, which is a separate analysis that we also offer. A typical gene-centric metagenomic study proceeds as follows:
We are eager to help you with your metagenomic analysis. Simply configure the analysis to your preferences, upload your raw sequences and metadata, and we can handle it from there. ▸ Overview of provided analysis
This analysis starts with raw short-read Illumina metagenomic sequences, which are provided by the client and typically obtained from a sequencing service provider. We deliver a summary report and key data products for presentations and downstream investigations. Main steps and deliverables:
In addition, we also provide a thorough Materials & Methods writeup for use in your publications. ▸ Input requirements
▸ Examples of data products
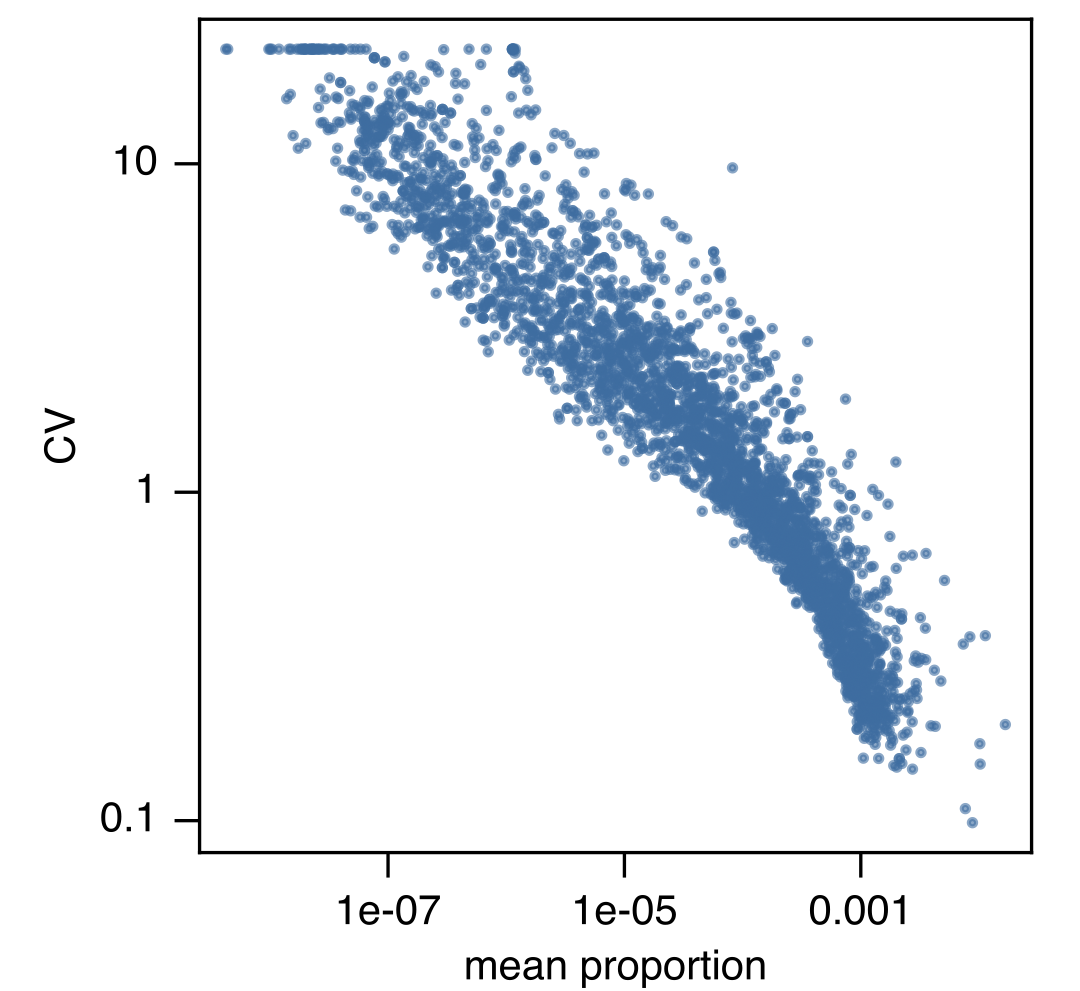
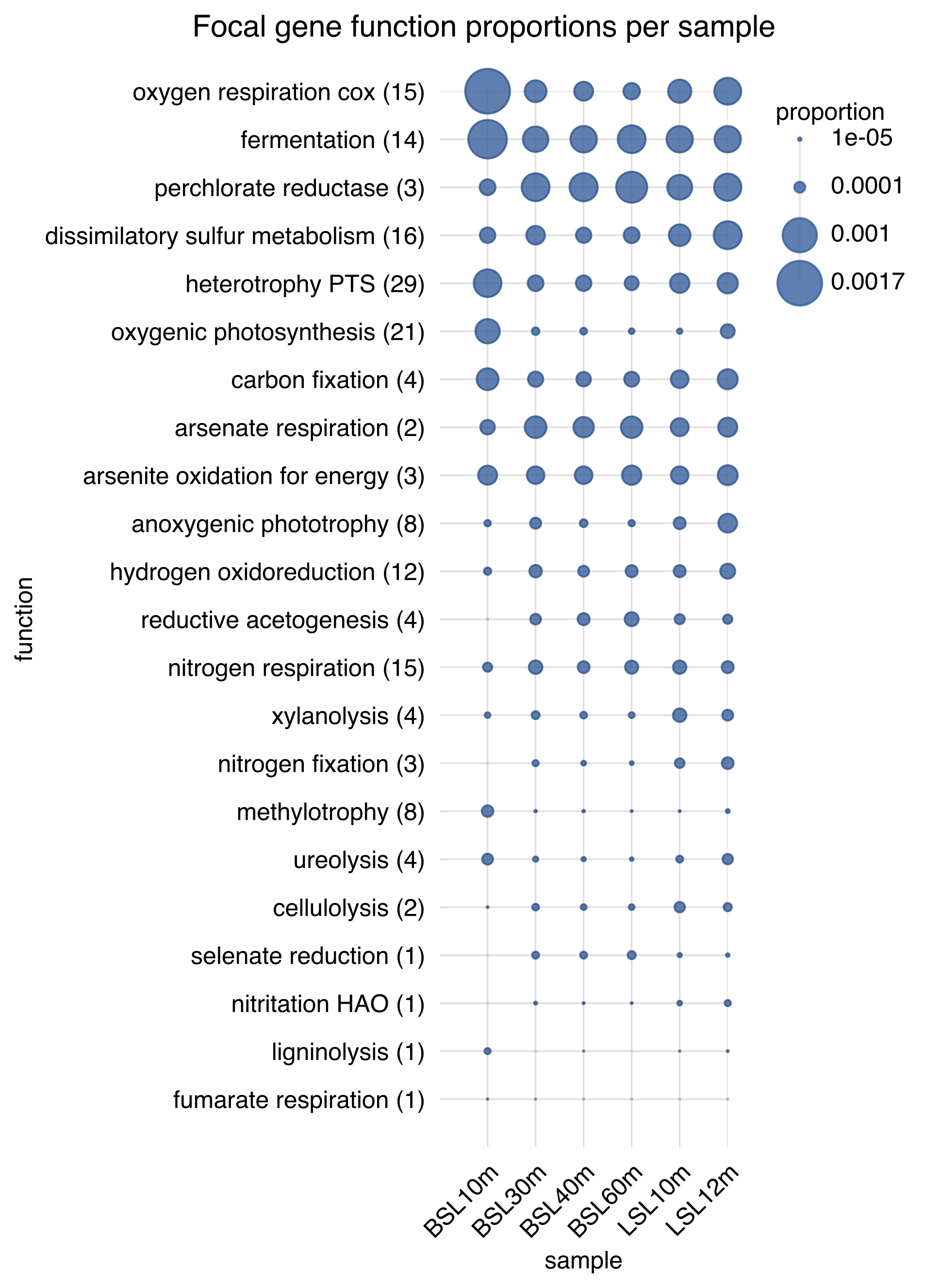
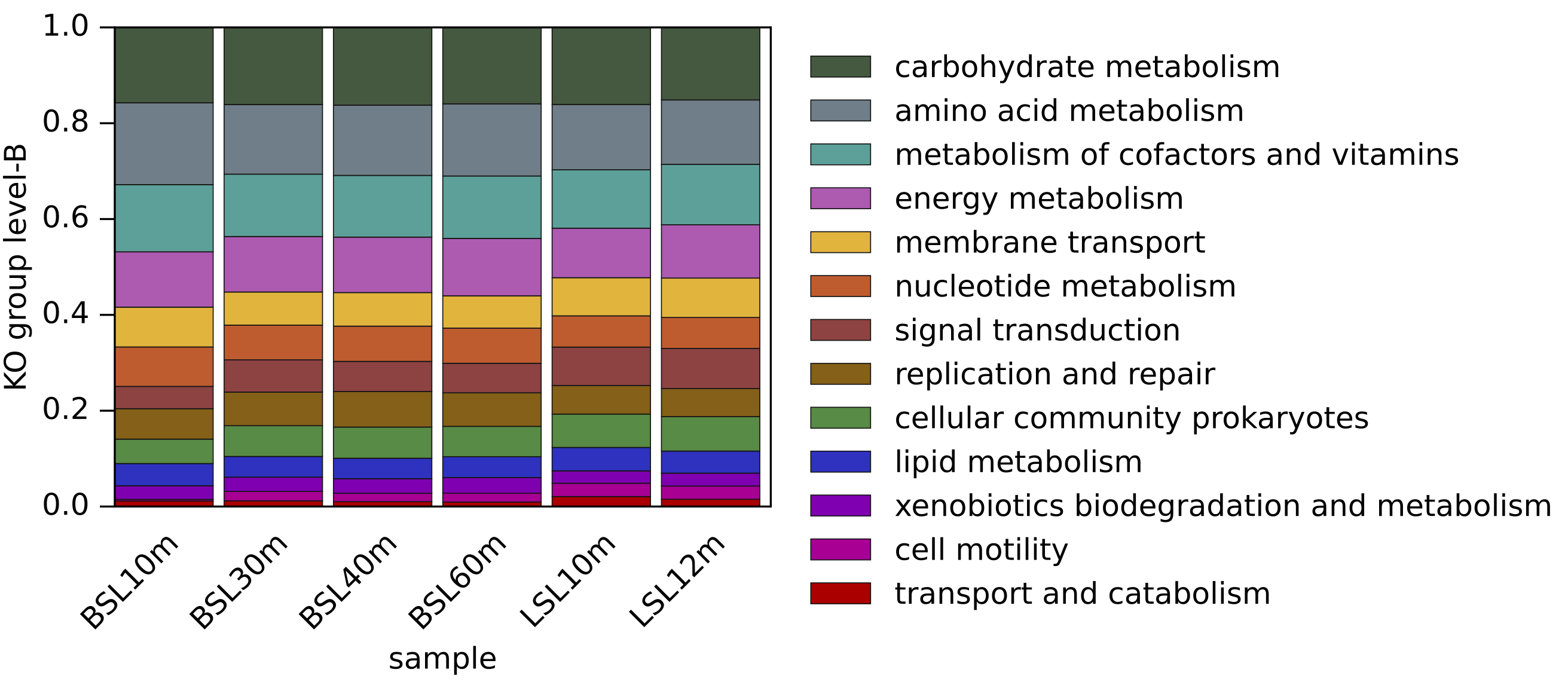
▸ Examples of generated figures
×
<
>
▸ Used 3rd party resources
Main databases and software used in this analysis:
▸ Relevant publications
▸ Price and billing
Price starts at $50 base + $5 per sample. Final price may differ depending on user settings, and will be available prior to order submission. Log in to see availability and payment modalities.
| ||||||||||
|
Policies About us Attributions |
Inferentus LLC PO box 554 Pleasant Hill OR, 97455, USA | |||||||||