Short-read genome-resolved metagenomics, including contig assembly, MAG binning, taxonomic identification, quality assessment, tree construction, functional annotation, estimation of relative abundances. The client provides raw shotgun metagenomic sequences and sample metadata.
Log in to see availability and payment modalities.
▾ General introduction
Metagenomic sequencing is a powerful technique in modern microbiology, in which random DNA fragments obtained from a biological sample are sequenced and analyzed.
These DNA fragments generally originate from many different taxa and many different genomic regions, thus offering a deep view of a microbial community.
Genome-resolved metagenomics is an increasingly popular variant whose goal it is to reconstruct the genomes of resident microbes by "binning" together sequences putatively originating from the same population.
The possibility of constructing such metagenome-assembled genomes (MAGs) is revolutionizing the field of microbiology, as it dramatically reduces the need for laborious culturing and yields detailed insight into the metabolic structure of complex microbial communities.
Genome resolved metagenomics has been used to recover novel MAGs from virtually every ecosystem, ranging from soils and the deep subsurface to the open ocean and animal guts.
In fact the recovery of MAGs has enabled the discovery of entire new phyla, dramatically expanding our view of extant microbial diversity.
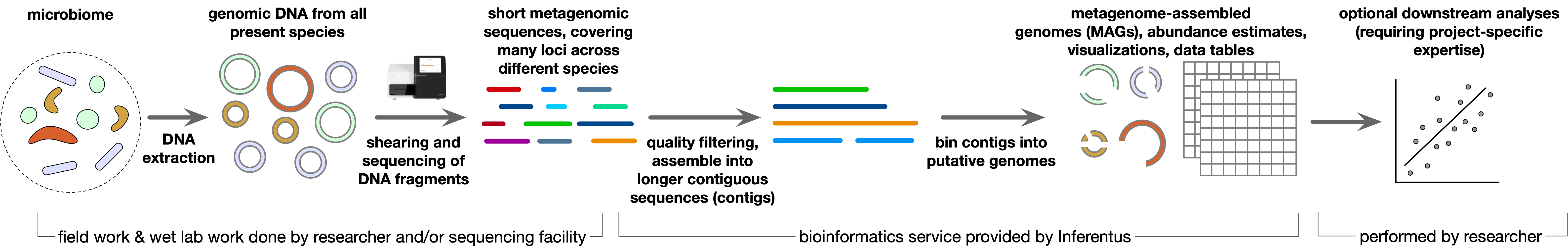
A typical genome-resolved metagenomic study proceeds as follows:
Collection of small amounts (<1 g) of material from each sample by the researcher.
Extraction of DNA from each sample using an in-house of commercial kit. This step is sometimes outsourced to an academic or commercial service provider.
DNA fragment size selection, library preparation and sequencing of the fragments. This step is commonly performed by an academic or commercial service provider. The most widespread technology is short read Illumina sequencing, which yields large numbers of sequences around 150-300 bp long.
Sequencing ultimately yields a separate set of DNA sequences for each sample, ranging from thousands to billions of sequences per sample, with each sequence covering some random part of some genome. These data are commonly stored in fastq files, which are delivered by the sequencing service provider to the researcher.
Computational analysis of the sequences, including trimming and removal of poor quality (i.e., likely erroneous) sequences, assembly of sequences into longer contiguous segments ("contigs"), estimation of contig abundances based on the number of reads mapped to each contig, binning contigs into MAG, and analyzing the MAGs.
We are eager to help you with your data analysis. Simply configure the analysis to your preferences, upload your raw sequence data and sample metadata, and we can handle it from there.
▸ Overview of provided analysis
This analysis starts with raw short-read Illumina metagenomic sequences, which are provided by the client and typically obtained from a sequencing service provider.
We deliver a summary report and key data products for presentations and downstream investigations.
Main steps and deliverables:
Basic quality filtering and trimming of sequences to improve overall data quality.
Assembly of reads into longer contiguous sequences (contigs).
Estimation of contig abundances based on the number of reads mapped to each contig.
Binning of contigs putatively originating from the same population, into metagenome-assembled genomes (MAGs).
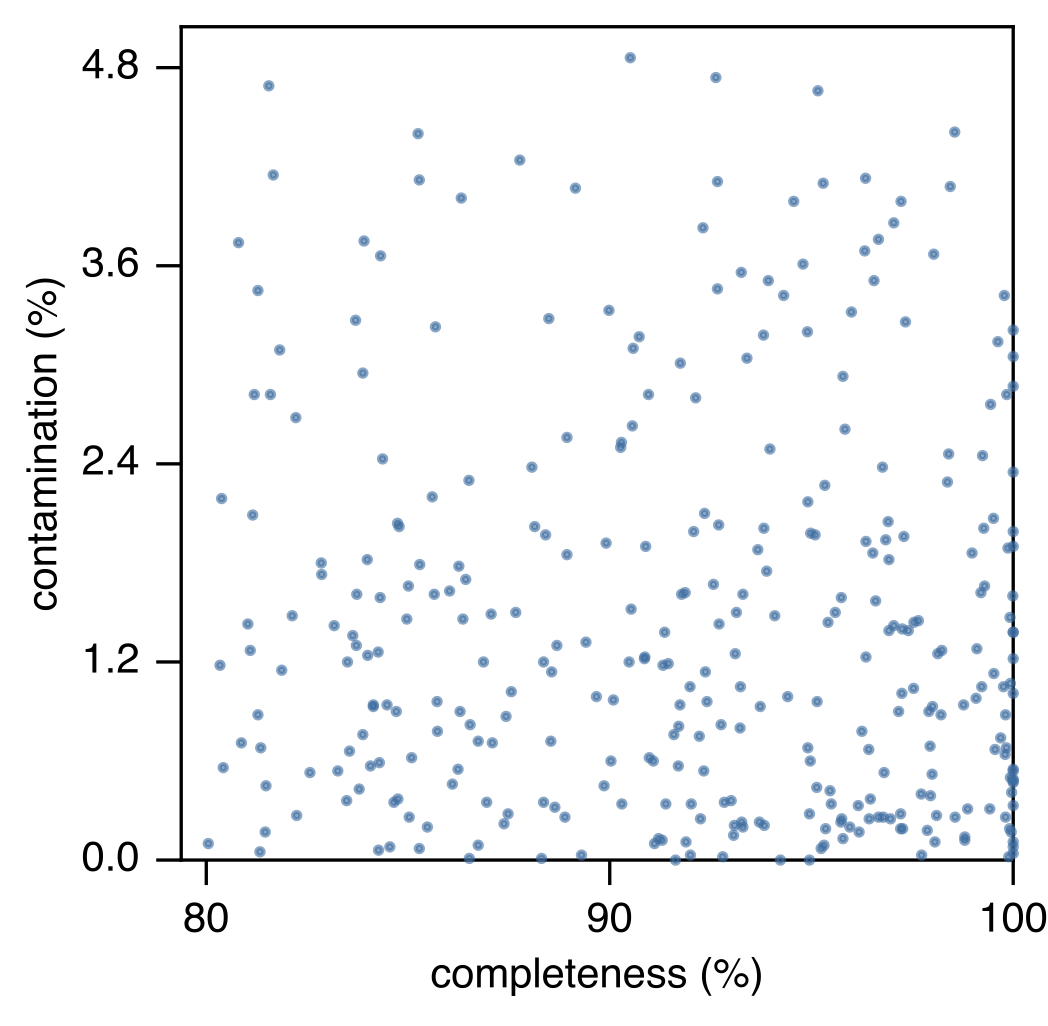
Assessment of MAG completeness and contamination levels.
Taxonomic classification of MAGs and phylogenetic tree construction based on single-copy "universal" genes.
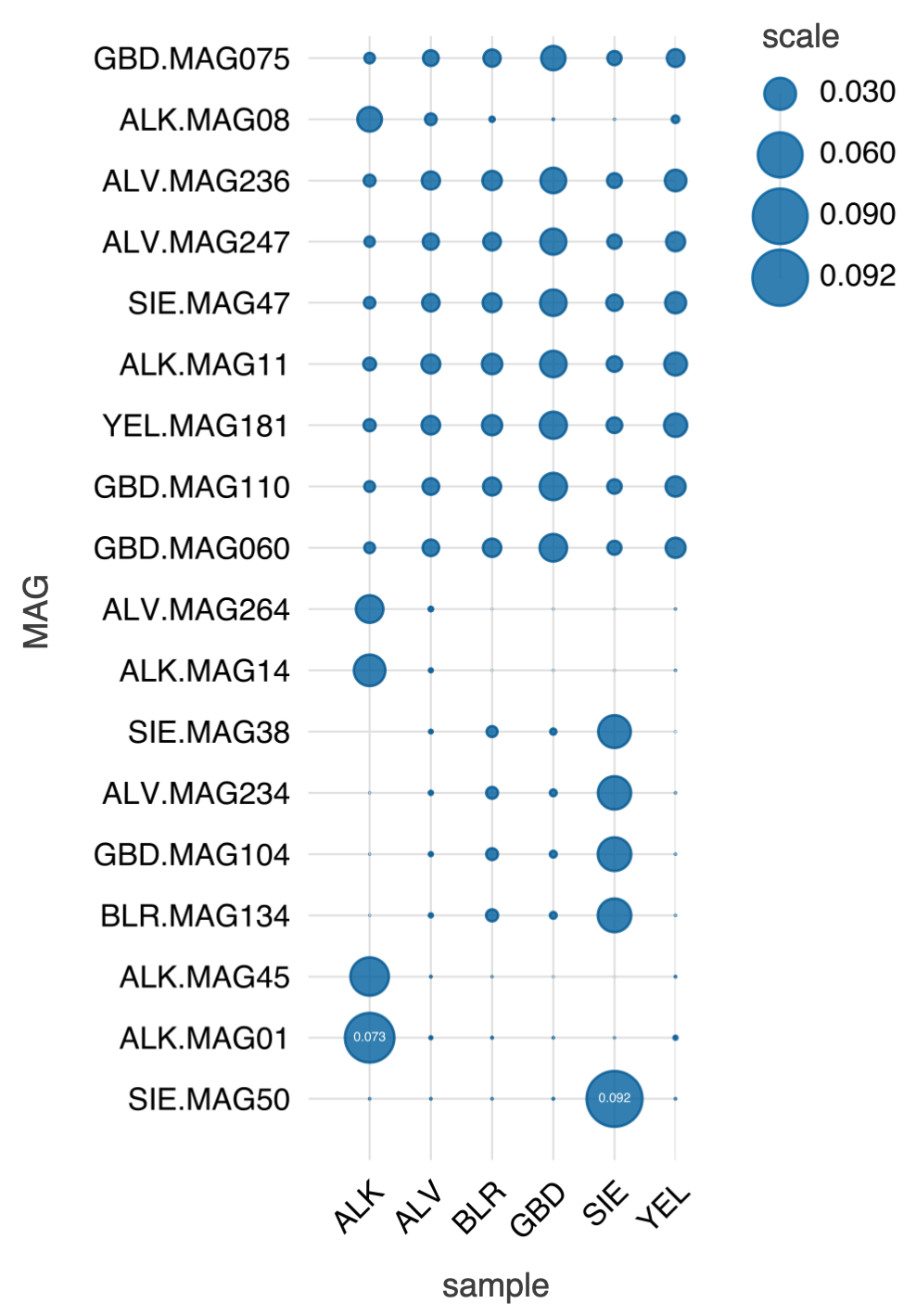
Estimation and visualization of MAG abundances in each sample based on the number of reads mapped to each MAG.
Detection and identification of protein-coding genes in each MAG, to determine each organism's metabolic potential.
In addition, we also provide a thorough Materials & Methods writeup for use in your publications.
▸ Examples of data products
MAG_ALK.MAG03.fasta.gz
Bacterial MAG (family Pyrinomonadaceae) recovered from hot springs, in compressed fasta file format.
MAG_tree_rooted_bacteria.tre
Multigene phylogeny of bacterial MAGs recovered from hot springs, in Newick file format.
MAG_proportions_by_sample.tsv
Table listing estimated relative abundances of MAGs in each sample.
MAG_taxonomies_qf.tsv
Table listing taxonomic classifications of recovered MAGs.
SGB_bin_members.tsv
Table listing species-level genome bins of closely related MAGs.
▸ Examples of generated figures
×<>
▸ Used 3rd party resources
Main databases and software used in this analysis:
Habibi-Soufi, H., Tran, D., Louca, S. (2024). Microbiology of Big Soda Lake, a multi-extreme meromictic volcanic crater lake in the Nevada desert. Environmental Microbiology 26:e16578
Hug, L. A., Baker, B. J., Anantharaman, K., Brown, C. T., Probst, A. J., Castelle, C. J. et al. (2016). A new view of the tree of life. Nature Microbiology 1:16048 EP
▸ Price and billing
Price starts at $50 base + $10 per sample. Final price may differ depending on user settings, and will be available prior to order submission. Log in to see availability and payment modalities.